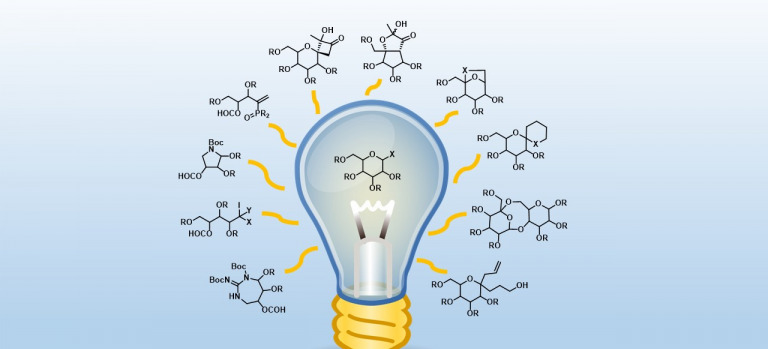
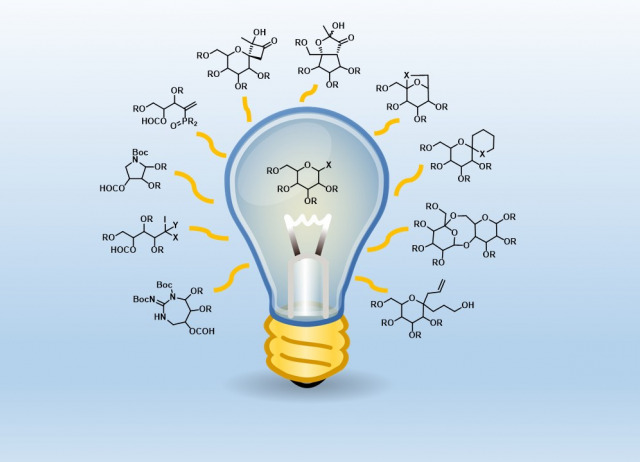
Síntesis de Productos Naturales
El grupo de Síntesis de Productos Naturales (SIPN), uno de los más antiguos del IPNA-CSIC, busca el desarrollo de nuevas metodologías sintéticas basadas en el uso de la luz para la modificación selectiva de biomoléculas con el fin de obtener productos de interés.
Perfil del grupo Síntesis de Productos Naturales en Digital.CSIC.
Presentación
Las líneas de investigación de nuestro grupo han evolucionado hasta centrarse en el campo de la metodología sintética, especialmente la química radicalaria. Así, se han estudiado ampliamente las reacciones de β-fragmentación (β-FRA) y transferencia intramolecular de hidrógeno (TIH) promovidas por radicales alcoxilo o amidilo, en presencia de reactivos de yodo hipervalente, como (diacetoxiyodo)benceno (DIB) o yodosilbenceno, y yodo con excelentes resultados, poniendo a punto una metodología, conocida como Reacción de Suárez, que caracteriza internacionalmente a nuestro grupo. Este protocolo implica la funcionalización regioselectiva de enlaces C-H de una manera altamente eficiente y sostenible.
Entre las aportaciones más novedosas e interesantes de las condiciones de Suárez destaca su aplicación a carbohidratos y esteroides con el fin de obtener una amplia batería de glicomiméticos y sintones quirales de difícil acceso por otras metodologías.
Actualmente, hemos extendido esta metodología a otro tipo de productos naturales enantiopuros accesibles y además, se estudian otros procesos bajo diversas condiciones de reacción, como en medio reductor, fotoquímicos u otras aplicaciones de los distintos sistemas redox de los halógenos, para la modificación quimioselectiva de moléculas orgánicas.
Líneas de investigación
Descarboxilación oxidativa y fotorrédox de ciclodextrinas
Esta línea de investigación se centra en el estudio de la reacción de β-fragmentación radicalaria inducida por la luz para promover la descarboxilación de mono- y di-ácidos carboxílicos en sistemas de polisacáridos cíclicos, como las ciclodextrinas.
Aplicación geobiomimética de la reactividad del yodo en el campo de la Síntesis Orgánica
En esta línea de investigación, nuestros esfuerzos se centran en una mejor comprensión de los fascinantes sistemas redox de yodo en diferentes entornos fisicoquímicos. Una característica relevante observada de estos sistemas redox ha sido la dinamicidad con la que transcurren las...
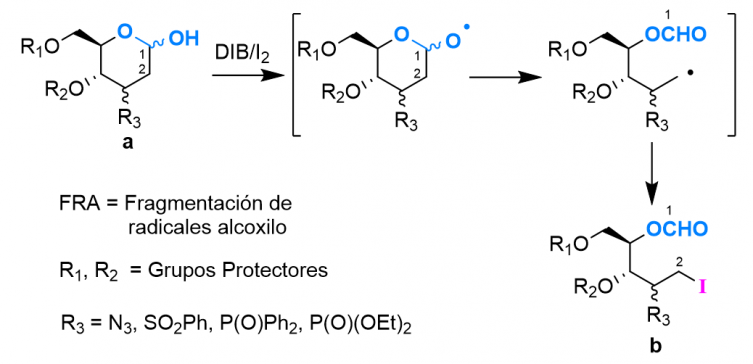
Fragmentación de radicales alcoxilo
La fragmentación radicalaria de carbohidratos1 es una línea de investigación desarrollada en nuestros laboratorios. En estudios previos2 realizados en nuestro grupo de trabajo, se ha encontrado que partiendo de carbohidratos (a) por β-fragmentación de radicales...
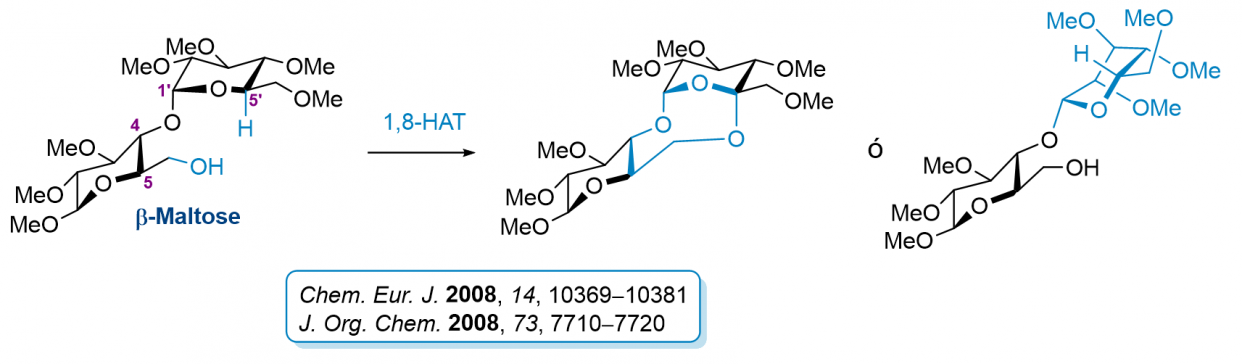
Procesos de Transferencia Intramolecular 1,8 en Sistemas Complejos de Carbohidratos
Los reacciones radicalarias de transferencia intramolecular de hidrógeno (TIH) que implican estados de transición de seis y siete miembros son muy frecuentes mientras que las que transcurren vía estados de transición superiores a siete miembros escasean. Recientemente, nuestro grupo ha sido...
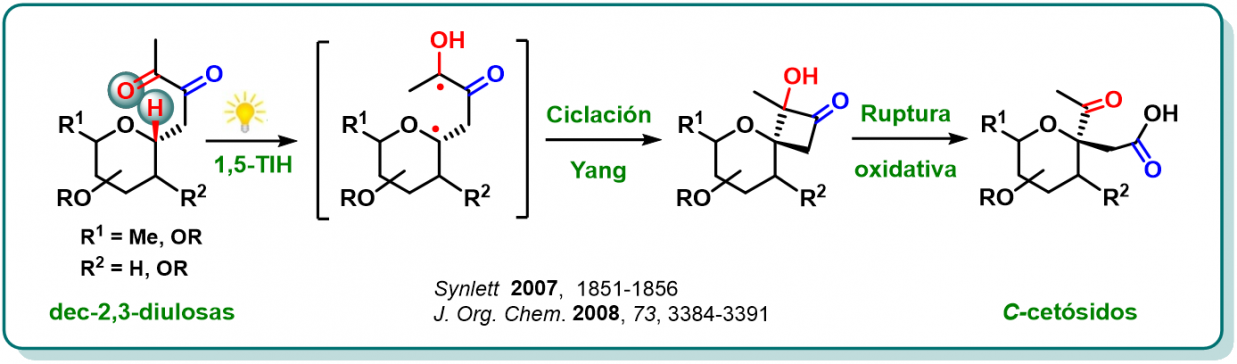
Reacciones Transferencia Intramolecular de Hidrógeno (TIH) promovidas por carbonilos fotoexcitados
Esta línea de investigación iniciada hace 10 años, se centra en el estudio de reacciones de TIH promovidas por carbonilos excitados fotoquímicamente.
Formación
Desarrollo de Nuevas Metodologías para la síntesis de inhibidores de glicosidasas y glicosiltransferasas y aplicación de la fotociclación de 1,2-dicetonas en el diseño de nuevos derivados polihidroxilados
Reacciones de Transferencia de Hidrógeno en Sistemas de Furanosas
Transferencia de átomos de Hidrógeno promovida por excitación fotoquímica de 1,2-dicetonas
Síntesis de Compuestos Organofosforados Altamente Funcionalizados, mediante Fragmentación de Radicales Alcoxilo Anóméricos de Carbohidratos. Aplicaciones Sintéticas
Desarrollo de nuevas metodologías para la síntesis de azepanos
TFG | Efectos del ácido glutámico y tres derivados en respuesta a estrés hídrico aplicado sobre Vicia faba L.
TFG | Efecto del ácido glutámico y su tosil-derivado en el crecimiento y respuesta al déficit hídrico en judía (Vicia faba L.)
TESIS | Funcionalización Selectiva de Enlaces C(sp3)–H Alifáticos usando Amidas como Grupos Directores y Sistemas Reactivos Basados en Yodo Hipervalente/yodo: Formación de Enlaces C-N Mediante Transferencia Intramolecular de Átomos de Hidrógeno
Supervisión de contratos en prácticas
Dirección de Estudiantes Universitarios Extranjeros
Dirección de prácticas de Fin de Ciclo Formativo de Técnico de Grado Superior de Química Ambiental
MÁSTER | Nuevos Sustituyentes sobre Radicales Centrados en Nitrógeno. Estudio de su Reactividad y Aplicaciones en Síntesis Asimétrica
TESIS | Fragmentación radicalaria de alcoholes anoméricos de carbohidratos: Síntesis de Heterociclos Polihidroxilados y Evaluación de su Actividad Biológica
MÁSTER | Desarrollo de Nuevas Metodologías para la Síntesis de Azepanos
Financiación

Oler para creer | Actividades paralelas
Desarrollo y ejecución de las actividades paralelas vinculadas a la exposición 'Oler para creer. La química del olor'.
Inés Pérez Martín

Estudios mecanísticos y aplicaciones metodológicas de la N-glicosilación de guanidinas: el caso de la ramnosilación de argininas por la enzima bacteriana EarP (N-RHAGED)
Proyecto bajo la gestión de la Agencia Española de Investigación. Este proyecto se enmarca en el Programa Estatal para Impulsar la Investigación Científico-Técnica y su Transferencia, del Plan...
En Ejecución

Oler para creer. La química del olor
'Oler para creer. La química del olor' es una exposición que propone entrelazar ciencia, historia y tecnología para ofrecer experiencias olfativas que conecten con los visitantes de una forma...
En Ejecución

El volcán de Cumbre Vieja por tierra, mar y aire
El proyecto 'El volcán de Cumbre Vieja por tierra, mar y aire' se materializó en la exposición 'Ceniza y lava. Revelaciones científicas junto al volcán', una experiencia inmersiva que invitaba a...
Finalizado

Estudio exhaustivo de las defensas inducidas en plantas mediante la aplicación exógena de derivados fluorescentes de MSB, vitamina K3 y bisulfito sódico (VK3-BODIPY)
Finalizado

Radicales Libres en la Síntesis de Nuevos Glicomiméticos con Potencial Actividad Terapeútica
Finalizado

Preparación de Nuevas Estructuras de Ciclodextrinas para el Transporte de Fármacos
Finalizado

Aplicaciones Sintéticas de los Procesos de Transferencia Intramolecular de Hidrógeno en Sistemas de Carbohidratos. Obtención de Nuevos Sintones Quirales
Finalizado

Diseño de compuestos enantioméricamente puros utilizando nuevas metodologias basadas en C-, N-y O-radicales. Aplicación a la síntesis de sustancias con actividad biológica
Finalizado

Reacciones de Transferencia Intramolecular de Hidrógeno sobre Carbohidratos. Nuevas Metodologías para la Síntesis de Compuestos Potencialmente Bioactivos
Finalizado
Personal
Elisa Isabel De León Alonso

María del Sol Rodríguez Morales

Concepción González Martín
Antonio Jesús Herrera González

María Ángeles Martín Hernández

Inés Pérez Martín

Esther María Martínez González

Nieves Rodríguez Paz

Andrés González Santana

Ricardo Guillermo Álvarez




Publicaciones
Synthesis of Chiral Polyhydroxylated Benzimidazoles by a Tandem Radical Fragmentation/Cyclization Reaction: A Straight Avenue to Fused Aromatic-Carbohydrate Hybrids
The synthesis of benzimidazole-fused iminosugars through a tandem β-fragmentation-intramolecular cyclization reaction is described. The use of the benzimidazole ring as the internal nucleophile and the use of phenyliodosophthalate (PhI(Phth)), a new metal-free and low toxic hypervalent iodine reagent, are the most remarkable novelties of this synthetic strategy. With this approach, we have demonstrated the usefulness of the fragmentation of anomeric alkoxyl radicals promoted by the PhI(Phth)/I system for the preparation of new compounds with potential interest for both medicinal and synthetic chemists.
André-Joyaux, Emy; Santana, Andrés G.; González Martín, Concepción C.
Reductive Radical Cascades Triggered by Alkoxyl Radicals in the β-Cyclodextrin Framework
The generation and fate of 2,3,6-icosa-O-methyl-β-cyclomaltoheptaos-6-O-yl radical under reductive conditions is described. Two radical cascade reactions are involved: the main one is triggered by a 1,8-HAT of the hydrogen at 5C. The radical can reach the anomeric hydrogen at 1C three sugar units ahead using a six-step sequence. The different hydrogen donor ability of the group 14 hydrides permits one to selectively stop the cascade at 5C, 2C, and 4C to obtain β-CD with a β-l-Idop unit, acyclic hepta-, and hexa-saccharide structures, respectively.
León, Elisa I.; Martín, Ángeles; Pérez-Martín, Inés; Suárez, Ernesto
Chemoselective Intramolecular Functionalization of Methyl Groups in Nonconstrained Molecules Promoted by N-Iodosulfonamides
Mechanistic evidence observed in Hofmann–Löffler–Freytag-type reactions has been crucial to achieve the chemoselective functionalization of methyl groups under mild conditions. Radical-mediated methyl iodination and subsequent oxidative deiodination are the key steps in this functionalization, where iodine chemistry has a pivotal role on the formation of the C–N bond. The concepts of single hydrogen atom transfer (SHAT) and multiple hydrogen atom transfer (MHAT) are introduced to describe the observed chemoselectivity.
Paz, Nieves R.; Rodríguez Sosa, Dionisio; Valdés, Haydée; Marticorena, Ricardo; Melián, Daniel; Copano, Belén; González Martín, Concepción C.; Herrera, Antonio J.
Easy access to modified cyclodextrins by an intramolecular radical approach
A simple method to modify the primary face of cyclodextrins (CDs) is described. The 6I‐O‐yl radical of α‐, β‐, and γ‐CDs regioselectively abstracts the H5II, located in the adjacent D‐glucose unit, by an intramolecular 1,8‐hydrogen‐atom‐transfer reaction through a geometrically restricted nine‐membered transition state to give a stable 1,3,5‐trioxocane ring. The reaction has been extended to the 1,4‐diols of α‐ and β‐CD to give the corresponding bis(trioxocane)s. The C2‐symmetric bis(trioxocane) corresponding to the α‐CD is a stable crystalline solid whose structure was confirmed by X‐ray diffraction analysis. The calculated geometric parameters confirm that the primary face is severely distorted toward a narrower elliptical shape for this rim.
Álvarez-Dorta, Dimitri; León, Elisa I.; Kennedy, Alan R.; Martín, Ángeles; Pérez-Martín, Inés; Suárez, Ernesto
Fragmentation of carbohydrate anomeric alkoxyl radicals: Synthesis of chiral polyhydroxylated β-iodo- and alkenylorganophosphorus(V) compounds
A direct approach to β‐iodophosphonates and β‐iodophosphine oxides from 2,3‐dideoxy‐3‐phosphoryl carbohydrate derivatives has been achieved by using the anomeric alkoxyl radical 1,2‐fragmentation protocol. The reaction has been conducted on carbohydrate derivatives under mild conditions with (diacetoxyiodo)benzene and molecular iodine. Subsequent dehydroiodination afforded the corresponding vinylphosphonates and vinylphosphine oxides.
Hernández-Guerra, Daniel; Rodríguez Morales, María S.; Suárez, Ernesto
Sequential Norrish type II photoelimination and intramolecular aldol cyclization of α-diketones: Synthesis of polyhydroxylated cyclopentitols by ring contraction of hexopyranose carbohydrate derivatives
The excitation of the innermost carbonyl of nono‐2,3‐diulose derivatives by irradiation with visible‐light initiates a sequential Norrish type II photoelimination and aldol cyclization process that finally gives polyfunctionalized cyclopentitols. The rearrangement has been confirmed by the isolation of stable acyclic photoenol intermediates that can be independently cyclized by a thermal 5‐(enolexo)‐exo‐trig uncatalyzed aldol reaction with high diastereoselectivity. In this last step, the large deuterium kinetic isotope effect found for the 1,5‐hydrogen atom transfer seems to indicate that the aldol reaction runs through a concerted pericyclic mechanism. Owing to the ready availability of pyranose sugars of various configurations, this protocol has been used to study the influence of pyranose ring‐substituents on the diastereoselectivity of the aldol cyclization reaction. In contrast with other pyranose ring contraction methodologies no transition‐metal reagents are needed and the sequential rearrangement occurs simply by using visible light and moderate heating (0 to 60 °C).
Álvarez‐Dorta, Dimitri; León, Elisa I.; Kennedy, Alan R.; Martín, Ángeles; Pérez-Martín, Inés; Riesco-Fagundo, Concepción; Suárez, Ernesto
Concepción González Martín

Datos de contacto
Noticias/Blog
- 21 Abril 2026
- 20 Febrero 2026
- 27 Diciembre 2023
- 20 Agosto 2019
Otros grupos de investigación